
Alzheimer’s disease (AD) is the most common form of dementia. It primarily affects adults over the age of sixty. AD affects the brain by destroying neurons—the basic components of the brain. Neurons are the chief type of cell destroyed by Alzheimer’s disease. Alzheimer’s disease leads to nerve cell death and tissue loss throughout the brain. Over time, the brain shrinks dramatically, affecting nearly all its functions. The real work of the brain goes on in individual cells. An adult brain contains about 100 billion nerve cells, or neurons, with branches that connect at more than 100 trillion points. Scientists call this dense, branching network a “neuron forest.” Signals traveling through individual nerve cells as tiny electrical charges move through this dense neuron forest to form the basis of memories, thoughts, and feelings. Nerve cells connect to one another at synapses. When a charge reaches a synapse, it may trigger release of tiny bursts of chemicals called neurotransmitters. The neuro-transmitters travel across the synapse, carrying signals to other cells. Scientists have identified dozens of neurotransmitters. Alzheimer’s disease disrupts both the way electrical charges travel within cells and the activity of neurotransmitters.
In advanced AD, massive cell loss changes the whole brain as follows:
- Thecortex shrivels up, damaging areas involved in thinking, planning and remembering;
- Shrinkage is especially severe in thehippocampus, an area of the cortex that plays a key role in formation of new memories;
- Ventricles(fluid-filled spaces within the brain) grow larger.
Many of the devastating effects of Alzheimer’s disease are visible when brain tissue is examined under the microscope. Alzheimer’s tissue has many fewer nerve cells and synapses than a healthy brain. In addition, plaques (abnormal clusters of protein fragments), build up between nerve cells. Finally, dead and dying nerve cells contain tangles, which are made up of twisted strands of another protein.
Scientists are not absolutely sure what causes cell death and tissue loss in the Alzheimer’s brain, but plaques and tangles are prime suspects.
- Amyloid Plaques: Plaques form when protein pieces called beta-amyloid clump together (beta-amyloid comes from a larger protein found in the fatty membrane surrounding nerve cells). Beta-amyloid is chemically “sticky” and gradually builds up into plaques. Many experts believe the most damaging form of beta-amyloid may be clumps of small pieces rather than the plaques themselves, because the small clumps may block cell-to-cell signaling at synapses. The clumps may also activate immune system cells that trigger inflammation and devour disabled cells. These plaques are believed to form early in the disease process, before neurons begin to die and the symptoms of memory loss and dementia become apparent.
- Neurofibrillary Tangles: Neurofibrillary refers to tiny filaments or fibers inside nerve cells. Neurofibrillary tangles form when threads of a protein called tau begin to twist. Without sufficient support, the twisted cell structure collapses. Tangles ultimately destroy a vital cell transport system made of proteins. In the healthy brain, the cell transport system isorganized in orderly parallel strandslike railroad tracks. Food molecules, cell parts and other key materials travel along these “tracks.”The tau protein helps the tracks stay straight. When tangles form, the tracks can no longer stay straight. The tracks fall apart and disintegrate, restricting the movement of nutrients and other essential supplies through the brain cells, which eventually die.
- Mis-folded Proteins:
Protein folding is the process by which a protein structure assumes its functional shape or conformation. It is the physical process by which apolypeptide folds into its characteristic and functional three-dimensional structure from random coil. Each protein exists as an unfolded polypeptide or random coil when translated from a sequence of mRNA to a linear chain of amino acids. This polypeptide lacks any stable (long-lasting) three-dimensional structure (the left hand side of the first figure). Amino acids interact with each other to produce a well-defined three-dimensional structure, the folded protein (the right hand side of the figure), known as the native state. The resulting three-dimensional structure is determined by the amino acid sequence (Anfinsen’s dogma).[2] Experiments [3] beginning in the 1980s indicate the codon for an amino acid can also influence protein structure.
The correct three-dimensional structure is essential to function, although some parts of functional proteins may remain unfolded,[4] so that protein dynamics is important. Failure to fold into native structure generally produces inactive proteins, but in some instances misfolded proteins have modified or toxic functionality. Several neurodegenerative and other diseases are believed to result from the accumulation of amyloid fibrils formed by misfolded proteins (as shown below).Many allergies are caused by incorrect folding of some proteins, for the immune system does not produce antibodies for certain protein structures.
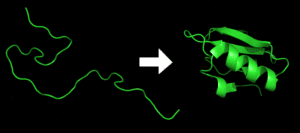
Protein before and after folding.
Aggregated proteins are associated with prion-related illnesses such as Creutzfeldt-Jakob disease, bovine spongiform encephalopathy (mad cow disease), amyloid-related illnesses such as Alzheimer’s disease and familial amyloid cardiomyopathy or polyneuropathy, as well as intracytoplasmic aggregation diseases such as Huntington’s and Parkinson’s disease.These age onset degenerative diseases are associated with the aggregation of misfolded proteins into insoluble, extracellular aggregates and/or intracellular inclusions including cross-beta sheet amyloid fibrils. While it is not completely clear whether the aggregates are the cause or merely a reflection of the loss of protein homeostasis, the balance between synthesis, folding, aggregation and protein turnover, the recent European Medicines Agency approval of Tafamidis or Vyndaqel (a kinetic stabilizer of tetrameric transthyretin) for the treatment of the transthyretin amyloid diseases suggests that it is the process of amyloid fibril formation and not the fibrils themselves that causes the degeneration of post-mitotic tissue in human amyloid diseases.[18] Misfolding and excessive degradation instead of folding and function leads to a number of proteopathy diseases such as antitrypsin-associatedemphysema, cystic fibrosis and the lysosomal storage diseases, where loss of function is the origin of the disorder. While protein replacement therapy has historically been used to correct the latter disorders, an emerging approach is to use pharmaceutical chaperones to fold mutated proteins to render them functional.
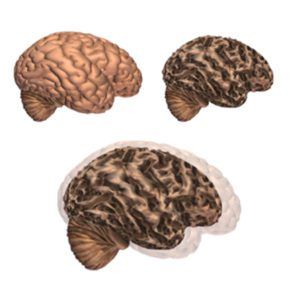
Alzheimer’s disease leads to nerve cell death and tissue loss throughout the brain. Over time, the brain shrinks dramatically, affecting nearly all its functions.
These images show:
- A brain without the disease
- A brain with advanced Alzheimer’s
More brain changes
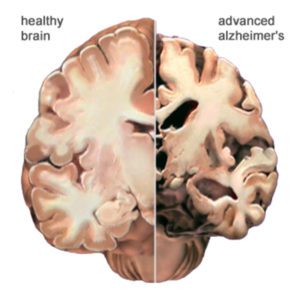
Here is another view of how massive cell loss changes the whole brain in advanced Alzheimer’s disease. This slide shows a crosswise “slice” through the middle of the brain between the ears.
In the Alzheimer’s brain:
- The cortex shrivels up, damaging areas involved in thinking, planning and remembering.
- Shrinkage is especially severe in the hippocampus, an area of the cortex that plays a key role in formation of new memories.
- Ventricles (fluid-filled spaces within the brain) grow larger.
EXERCISE AND ALZHEIMER’S DISEASE
A number of recent clinical trials have shown numerous benefits for AD sufferers from physical exercise. Physical activity was shown to improve mood, memory and ability to think for participants in three new studies.One study found that intense aerobic exercise improves blood flow to key areas of the brain, and appears to reduce the tau protein tangles that are a hallmark of Alzheimer’s disease. According to one of the stdy leaders, Laura Baker (cognitive neuroscientist at Wake Forest School of Medicine in Winston-Salem, N.C.):
“Blood flow decreases in those areas for all of us with age, and yet exercise increased it.” “It seems to me we’re changing aging-related effects, and we may be changing Alzheimer’s-related effects, both with exercise.”
The three new studies “give us information about living better with the disease,” said Heather Snyder, director of medical and scientific operations for the Alzheimer’s Association.”Physical exercise is potentially beneficial to people who are living with Alzheimer’s today,” Snyder said. “Even once you have cognitive impairment, there’s still a benefit to physical activity.”
Earlier research has shown that exercise can improve the ability to think in healthy adults, so Baker and her colleagues turned to people with mild impairment to see if physical activity would help them, too. The 65 people in Baker’s study were 55 to 89 years old and had not been exercising beforehand. They also had prediabetes, which can increase risk of developing Alzheimer’s disease.Participants were randomly assigned to one of two groups for six months. The first group performed stretching exercises that did not raise their heart rate much, while the second group had to perform at least 45 minutes of high-intensity aerobics four times a week.
The aerobics group had to stay within 75 percent to 85 percent of their maximum heart rate for at least 30 minutes of their workout, which most often took place on a treadmill. “For our typical 70-year-old, that means a heart rate of at least 130 beats per minute,” Baker said.Ninety-two percent of people stuck to the exercise program, and wound up with improved fitness and better blood sugar levels, researchers found.
More important, MRI brain scans revealed that blood flow had significantly increased to the memory and processing centers of participants’ brains, with a corresponding improvement in their ability to plan, organize and pay attention.Tests using cerebro-spinal fluid samples drawn from the patients also showed a significant reduction in tau protein tangles, with the effect most pronounced in those older than 70.”These findings are important because they strongly suggest a potent lifestyle intervention such as aerobic exercise can impact Alzheimer’s-related changes in the brain,” Baker said. “No currently approved medication can rival these effects.”
In another clinical trial, 200 people between ages 50 and 90 with Alzheimer’s were randomly assigned to either an aerobic exercise program or a control group that performed no extra exercise. The folks who exercised were asked to reach a target intensity of 70 percent to 80 percent of their maximum heart rate.The Danish researchers found that those who exercised suffered from fewer mood problems such as anxiety, irritability and depression. The people who exercised most often and most vigorously also achieved significant improvements in mental speed and attention.
The third clinical trial took place in Canada and involved 71 people between ages 56 and 96 who had suffered ministrokes, diminishing their ability to think and remember. Half were assigned to a group that took part in regular aerobics classes.The researchers found that participants who took aerobics significantly improved their memory and selective attention, compared with those not asked to exercise regularly.
- 2/3rds of AMERICA’S ALZHEIMER’S SUFFERERS ARE WOMEN
- Two-thirds of Americans suffering from Alzheimer’s are women.
- Women with memory problems that may signal early Alzheimer’s descend into dementia twice as fast as men, according to recent research.
- Women in their 60s are twice as likely to develop Alzheimer’s as they are to get breast cancer (Alzheimer’s Association).
Researchers suspect that the pronounced differences between men and women and Alzheimer’s has something to do with the biology of the brain. Experts say it may relate to the complicated interaction between genetics, hormones and the way the brain develops. Studies have found a number of differences between gender, including findings that women are more likely than men to suffer from depression (a known risk factor for Alzheimer’s disease) and that women are more vulnerable to stress (also a risk factor for Alzheimer’s).(1) It has also been found women have more of the brain-clogging protein called amyloid that is a hallmark of Alzheimer’s.
A recent study evaluated 1,000 people: 273 normal people, 557 with mild cognitive impairment and 145 who had diagnosed Alzheimer’s. The study confirmed that women have more amyloid in the brain than men even when other factors were adjusted for.(1)
(1) Presented along with a series of studies presented July 21, 2015 at the Alzheimer’s Association International Conference (Dr. Michael Weiner, et al, University of California at San Francisco).
- BDNF AND ALZHEIMER’S DISEASE
Brain-derived neurotrophic factor (BDNF) has been described by some experts as “Miracle-Gro for the brain” due to its properties of strengthening existing neurons and helping new neurons grow. Recent evidence suggests that amyloid-beta associated neurotoxicity may be a consequence of a deficiency of BDNF. Several studies indicate that the cortex and hippocampus, areas of the brain associated with learning and memory, exhibit both extensive amyloid pathology and decreased levels of BDNF in AD (Phillips et al., 1991; Connor et al., 1997; Ferrer et al., 1999; Hock et al., 2000; Holsinger et al., 2000; Garzon et al., 2002; Peng et al., 2005). Interestingly, BDNF protein levels are significantly decreased in preclinical and early stages of AD, and this reduction correlates with clinical neuropsychological scores (Peng et al., 2005). Since BDNF is critical for neuronal survival and function (Siegel and Chauhan, 2000; Mufson et al., 2007) and for synaptic plasticity and learning and memory (Korte et al., 1995; Patterson et al., 1996; Lu, 2003; Bramham and Messaoudi, 2005; Nagahara et al., 2009), which are compromised in AD, it is important to understand which Aβ species drive the reduction of BDNF in AD. In vitro data demonstrate that soluble forms of Aβ decrease BDNF mRNA expression and compromise BDNF intracellular signaling in both primary rat neurons and human neuroblastoma cells (Tong et al., 2001,2004;Garzon and Fahnestock, 2007). Therefore, amyloid-induced neurodegeneration may be a consequence of reduced BDNF. However, whether Aβ assemblies downregulate BDNF in vivo, and which Aβ assembly state is responsible for BDNF downregulation, have not been elucidated. In this study, we measured levels of BDNF mRNA and Aβ42/Aβ40 ratios and characterized the state of Aβ in three different transgenic mouse models of AD (Table 1) containing mutations in APP, two of these in combination with PS-1 mutations, and in a mouse model of Down syndrome (segmental trisomy 16) containing an additional copy of App.
Neurobiol Dis. 2008 Sep;31(3):316-26. doi: 10.1016/j.nbd.2008.05.012. Epub 2008 Jul 15.
Protective effect of BDNF against beta-amyloid induced neurotoxicity in vitro and in vivo in rats.
Arancibia S1, Silhol M, Moulière F, Meffre J, Höllinger I, Maurice T, Tapia-Arancibia L.
We examined the potential protective effect of BDNF against beta-amyloid-induced neurotoxicity in vitro and in vivo in rats. In neuronal cultures, BDNF had specific and dose-response protective effects on neuronal toxicity induced by Abeta(1-42) and Abeta(25-35). It completely reversed the toxic action induced by Abeta(1-42) and partially that induced by Abeta(25-35). These effects involved TrkB receptor activation since they were inhibited by K252a. Catalytic BDNF receptors (TrkB.FL) were localized in vitro in cortical neurons (mRNA and protein). In in vivo experiments, Abeta(25-35) was administered into the indusiumgriseum or the third ventricle and several parameters were measured 7 days later to evaluate potential Abeta(25-35)/BDNF interactions, i.e. local measurement of BDNF release, number of hippocampal hilar cells expressing SRIH mRNA and assessment of the corpus callosum damage (morphological examination, pyknotic nuclei counting and axon labeling with anti-MBP antibody). We conclude that BDNF possesses neuroprotective properties against toxic effects of Abeta peptides.
J Neurochem. 2004 Feb;88(4):1010-8.
Regulation of beta-amyloid precursor protein expression by brain-derived neurotrophic factor involves activation of both the Ras and phosphatidylinositide 3-kinase signalling pathways.
Ruiz-León Y1, Pascual A.
Brain-derived neurotrophic factor (BDNF) stimulates beta-amyloid precursor protein (APP) promoter activity by a Ras-dependent mechanism in TrkB-expressing SH-SY5Y cells. To determine the signalling pathways involved in the BDNF-induced response, we have analysed the ability of TrkB mutated forms to mediate promoter stimulation. Brain-derived neurotrophic factor causes a significant induction of promoter activity and mutation K540R in the active site of TrkB completely abolishes the neurotrophin-induced response. A substitution of the Y484 residue by phenylalanine, which blocks binding of Shc, reduces the activation of APP promoter by BDNF by approximately 50% whereas mutation Y785P, which blocks binding of phospholipase C gamma, does not affect the response. In addition, the phosphatidylinositide 3-kinase (PI3K)-specific inhibitors wortmannin and LY294002 reduced BDNF-induced activation. In agreement with a participation of both Ras/MAPK- and PI3K/Akt-mediated mechanisms, transient expression of constitutive active forms of Ras, PI3K and other components of both signalling pathways led to a significant increase of APP promoter activity. Furthermore, the stimulation of the APP promoter by BDNF was completely precluded by expression of dominant-negative forms of Ras and PI3K effectors. Taken together, our results suggest that simultaneous activation of at least two signalling pathways, Ras/MAPK and PI3K/Akt, is necessary to mediate a full activation of the APP promoter by BDNF.
J Neurosci. 2009 Jul 22;29(29):9321-9. doi: 10.1523/JNEUROSCI.4736-08.2009.
Decreased brain-derived neurotrophic factor depends on amyloid aggregation state in transgenic mouse models of Alzheimer’s disease.
Peng S1, Garzon DJ, Marchese M, Klein W, Ginsberg SD, Francis BM, Mount HT, Mufson EJ, Salehi A, Fahnestock M.
Downregulation of brain-derived neurotrophic factor (BDNF) in the cortex occurs early in the progression of Alzheimer’s disease (AD). Since BDNF plays a critical role in neuronal survival, synaptic plasticity, and memory, BDNF reduction may contribute to synaptic and cellular loss and memory deficits characteristic of AD. In vitro evidence suggests that amyloid-beta (A beta) contributes to BDNF downregulation in AD, but the specific A beta aggregation state responsible for this downregulation in vivo is unknown. In the present study, we examined cortical levels of BDNF mRNA in three different transgenic AD mouse models harboring mutations in APP resulting in A beta overproduction, and in a genetic mouse model of Down syndrome. Two of the three A beta transgenic strains (APP(NLh) and TgCRND8) exhibited significantly decreased cortical BDNF mRNA levels compared with wild-type mice, whereas neither the other strain (APP(swe)/PS-1) nor the Down syndrome mouse model (Ts65Dn) was affected. Only APP(NLh) and TgCRND8 mice expressed high A beta(42)/A beta(40) ratios and larger SDS-stable A beta oligomers (approximately 115 kDa). TgCRND8 mice exhibited downregulation of BDNF transcripts III and IV; transcript IV is also downregulated in AD. Furthermore, in all transgenic mouse strains, there was a correlation between levels of large oligomers, A beta(42)/A beta(40), and severity of BDNF decrease. These data show that the amount and species of A beta vary among transgenic mouse models of AD and are negatively correlated with BDNF levels. These findings also suggest that the effect of A beta on decreased BDNF expression is specific to the aggregation state of A beta and is dependent on large oligomers.
New clinical studies have shown that BDNF slows cognitive decline in pre-clinical Alzheimer’s disease.
Molecular Psychiatry advance online publication 7 October 2014; doi: 10.1038/mp.2014.123
APOE and BDNF polymorphisms moderate amyloid β-related cognitive decline in preclinical Alzheimer’s disease
Y Y Lim1,2,3, V L Villemagne1,4,5, S M Laws6,7,8, R H Pietrzak9, P J Snyder2,3, D Ames10,11, K A Ellis1,11, K Harrington1, A Rembach1, R N Martins6, C C Rowe4,5, C L Masters1 and P Maruff1,12
Received 19 May 2014; Revised 29 July 2014; Accepted 21 August 2014
Advance online publication 7 October 2014
Topof page
Abstract
Accumulation of β-amyloid (Aβ) in the brain is associated with memory decline in healthy individuals as a prelude to Alzheimer’s disease (AD). Genetic factors may moderate this decline. We examined the role of apolipoprotein E (ε4 carrier[ε4+], ε4 non-carrier[ε4−]) and brain-derived neurotrophic factor (BDNFVal/Val, BDNFMet) in the extent to which they moderate Aβ-related memory decline. Healthy adults (n=333, Mage=70 years) enrolled in the Australian Imaging, Biomarkers and Lifestyle study underwent Aβ neuroimaging. Neuropsychological assessments were conducted at baseline, 18-, 36- and 54-month follow-ups. Aβ positron emission tomography neuroimaging was used to classify participants as Aβ− or Aβ+. Relative to Aβ−ε4−, Aβ+ε4+ individuals showed significantly faster rates of cognitive decline over 54 months across all domains (d=0.40–1.22), while Aβ+ε4− individuals showed significantly faster decline only on verbal episodic memory (EM). There were no differences in rates of cognitive change between Aβ−ε4− and Aβ−ε4+ groups. Among Aβ+individuals, ε4+/BDNFMet participants showed a significantly faster rate of decline on verbal and visual EM, and language over 54 months compared with ε4−/BDNFVal/Val participants (d=0.90–1.02). At least two genetic loci affect the rate of Aβ-related cognitive decline. Aβ+ε4+/BDNFMet individuals can expect to show clinically significant memory impairment after 3 years, whereas Aβ+ε4+/BDNFVal/Val individuals can expect a similar degree of impairment after 10 years. Little decline over 54 months was observed in the Aβ− and Aβ+ ε4− groups, irrespective of BDNF status. These data raise important prognostic issues in managing preclinical AD, and should be considered in designing secondary preventative clinical trials.
- HEAT SHOCK PROTEINS (HSPs) AND ALZHEIMER’S DISEASE
Heat shock proteins (HSPs): HSPs are a family of proteins that are produced by cells in response to exposure to stressful conditions. They were first described in relation to heat shock but are now known to also be expressed during other stresses including exposure to cold, UV light, and during wound healing or tissue remodeling. Many members of this group perform chaperone function by stabilizing new proteins to ensure correct folding or by helping to refold proteins that were damaged by the cell stress. Recent studies are showing that HSPs can be neuro-protective in Alzheimer’s disease.
Int J Hyperthermia. 2005 Aug;21(5):421-31.
Stress proteins in Alzheimer’s disease.
Smith RC1, Rosen KM, Pola R, Magrané J.
Environmental and genetic conditions can cause proteins to misfold or to accumulate abnormally due to impaired clearance. The chaperones which include heat shock proteins, aid survival by preventing protein mis-folding and the formation of cytotoxic protein aggregates. An increasing number of studies point to important roles for molecular chaperones in the biology of neurodegenerative diseases. Heat shock proteins can suppress neurotoxicity in animal models of Parkinson’s and polyglutamine diseases, suggesting potential new therapeutic approaches in neurodegenerative disorders associated with abnormal protein folding and toxicity. Recent findings suggest that heat shock proteins can also be neuroprotective in Alzheimer’s disease, but this area of research remains largely unexplored. This paper will review the literature related to the role of heat shock proteins in Alzheimer’s disease.
Neuroscience. 2000;99(2):317-25.
Expression of amyloid precursor protein in human astrocytes in vitro: isoform-specific increases following heat shock.
Shepherd CE1, Bowes S, Parkinson D, Cambray-Deakin M, Pearson RC.
The beta-amyloid protein deposited in senile plaques and cerebral blood vessels in the Alzheimer’s disease brain is derived from the larger transmembrane spanning amyloid precursor protein. The present study investigates the effects of heat shock on the expression and processing of amyloid precursor protein in a normal human fetal astrocytic cell line CC2565 using reverse transcription-polymerase chain reaction, in situ hybridization histochemistry and western blot analysis. Heat shock led to an increase in the messenger RNA encoding Kunitz protease inhibitor isoforms of amyloid precursor protein, which peaked at 4h post-heat shock. This increase was confined to the messenger RNA encoding amyloid precursor protein-751, with a decrease in amyloid precursor protein-770 and no change in amyloid precursor protein-695. This shift in splicing was accompanied by a significant decrease in secreted amyloid precursor protein and an increase in beta-secretase processing within the cell. These findings demonstrate that astrocytes in vitro demonstrate a striking response to heat shock. This is unlikely to be due to a direct action on the promoter region of the gene, since the response is specific for one splice variant; amyloid precursor protein-751 messenger RNA. This increase in expression is further accompanied by a decrease in secretion of amyloid precursor protein, implying a shift in processing towards an intracellular route, possibly via the actions of the beta-secretase enzyme, which is known to be potentially amyloidogenic. Such a mechanism may contribute to amyloidosis in the intact brain in response to cellular stress, such as head injury.MolNeurobiol. 2007 Jun;35(3):203-16.
MolNeurobiol. 2007 Jun;35(3):203-16.
Heat shock proteins and amateur chaperones in amyloid-Beta accumulation and clearance in Alzheimer’s disease.
Wilhelmus MM1, de Waal RM, Verbeek MM.
The pathologic lesions of Alzheimer’s disease (AD) are characterized by accumulation of protein aggregates consisting of intracellular or extracellular misfolded proteins. The amyloid-beta (Abeta) protein accumulates extracellularly in senile plaques and cerebral amyloid angiopathy, whereas the hyperphosphorylated tau protein accumulates intracellularly as neurofibrillary tangles. “Professional chaperones”, such as the heat shock protein family, have a function in the prevention of protein misfolding and subsequent aggregation. “Amateur” chaperones, such as apolipoproteins and heparan sulfate proteoglycans, bind amyloidogenic proteins and may affect their aggregation process. Professional and amateur chaperones not only colocalize with the pathological lesions of AD, but may also be involved in conformational changes of Abeta, and in the clearance of Abeta from the brain via phagocytosis or active transport across the blood-brain barrier. Thus, both professional and amateur chaperones may be involved in the aggregation, accumulation, persistence, and clearance of Abeta and tau and in other Abeta-associated reactions such as inflammation associated with AD lesions, and may, therefore, serve as potential targets for therapeutic intervention.
——————————————————————————————————————————————————————–
APL (American Performance Labs) is a research group dedicated to the collection, analysis, and dissemination of published research and articles on the science of hyperthermia and the various applications, technologies and protocols for the use of hyperthermic conditioning.